
Why This Paper?
Weekly Study에서 두 번째로 다루어 볼 논문은 Exploring “dark-matter” protein folds using deep learning이다. 이 논문은 연구실 인턴을 하면서 듣게 된 protein fold space라는 개념에 대한 궁금증을 풀고, deep learning을 이용해서 어떤 방식으로 무엇을 연구하고 있는지 알기 위해서 선정했다.
About Paper
Exploring “dark-matter” protein folds using deep learning이라는 논문은 Cell Systems에 게재된 논문이다. 교신저자인 Bruno E. Correia 교수님은 스위스 로잔 연방 공과대학 소속으로 immunoprotein에 중점을 두어 protein design, drug discovery, bioengineering 분야의 연구를 계속 하고 계신다. 주요 저자들인 Zonder Harteveld와 Alexandra Van Hall-Beauvais도 역시 동일한 대학과 Swiss Institute of Bioinformatics (SIB)라는 곳에 소속되어 있고, AI-based protein engineering 분야의 논문을 계속 내고 있어서 이 분야에 대해 알아보는 데 좋은 논문으로 생각된다.
Main Points
1) De novo protein design은 진화에 의해 생성되지 않은 새로운 protein을 만들기 위해서 아직 기록되고 구별되지 않은 sequence & structure space를 탐색하는 것이다.
2) 주요한 문제는 target structure에 맞는 서열을 찾기 위해서 guide가 될 수 있는 “designable” structural template이 필요하다는 것인데, 저자들은 convolutional VAE (named Genesis)로 단백질 구조의 패턴을 학습하고, trRosetta와 함께 여러 fold에 대한 sequence를 design하였다.
3) Genesis는 native folds는 물론, “dark-matter” folds에 관해서도 일반화되게 distance and angle distribution을 잘 구성하였다 (proteases resistance를 이용해 안정성 검증).
4) Small neural network가 protein structural pattern을 효과적으로 학습하고, backbone design problem을 해결할 수 있음을 보였다.
Main Texts
Introduction
직관적으로 protein sequence space라는 것을 생각해보면, 만약 서열의 길이가 3일 경우 3차원 좌표계를 상상하고, 각 축에서 20가지의 amino acids 중 하나를 선택하여 찍게 되는 하나의 점이 바로 서열 3개의 protein sequence space 내의 하나의 protein sequence로 표현될 것이다. 실제로 기능하는 단백질들은 수 백에서 수 만의 amino acids를 이용해서 구성된다. 자연에서 일어나고 있는 evolution은 이러한 possible protein sequence space의 일부분을 조금씩 탐색하는 과정으로 생각할 수 있다. 그리고 대부분의 서열들은 유한한 개수의 protein folds 집합으로 분류될 수 있는 3차원 구조로 접히게 되는데, 이러한 folds의 집합을 protein fold space라고 볼 수 있다.
de novo protein design은 알려진 native fold space의 내부 혹은 그 밖에 정의된 3D conformation으로 잘 접힐 수 있는 novel sequences를 찾아가는 과정이다. 이러한 연구는 machine learning technique을 적극적으로 활용하고 있고, 많은 경우 two-step process로 진행된다.
1) protein fold is outlined and corresponding backbones are generated (backbone은 각 아미노산에서 $\alpha$-carbon으로 구성된 뼈대를 의미한다).
2) amino acid (aa) sequences are searched to fit the generated backbones.
물론 두 과정 각각에 대한 어려운 점들이 존재하고 있다. 먼저 backbone generation의 경우, 물리적으로 비현실적인 경우 실험에서 접힘이 불가능하다. 그래서 궁극적으로 20개의 natural a.a.를 가지고 효과적으로 3차 형태로 packing 될 수 있는 최적의 secondary structure configuration (구성)을 찾게 되는데, 이를 designable backbones이라 한다. Backbone의 designability가 높을수록 그 backbone에 맞는 더 다양하고 많은 energetically favorable sequence를 찾을 수 있지만, 이를 정량적으로 표시하고 평가하는 것이 매우 어렵다.
그리고 amino acid sequence generation의 측면에서는 최근 많은 발전이 이루어지고 있다. AlphaFold, RoseTTAfold 등의 서열로부터 구조를 매우 정확하게 예측하는 모델들은 deep neural networks (DNNs)의 발달과 함께 large-scale protein sequence & structural dataset이 이용 가능해졌기 때문에 가능했다. 실제로 transform-restrained Rosetta (trRosetta)나 AlphaFold (AF)의 신경망에서 target structure to sequence로 gradient backpropagation을 통한 학습을 진행하면, 우리는 structure prediction methods를 역으로 활용하여 fixed backbone으로부터 서열을 생성하는 protein design task를 풀 수 있게 된다. 특히 network를 거치면서 나오는 서열들은 전체적인 학습 데이터 상의 서열과 구조 관계 속에서 최적화를 거치게 되고, 의미상으로는 lowest energy를 가지면서도 다른 구조들과 비교해서 특정한 하나의 target structure로 잘 접힐 수 있는 sequence가 된다. 혹은 protein backbone을 structure-based context로 활용해서 서열을 생성하는 방법도 많이 이용되고 있다. ProteinMPNN에서 MPNN에 해당하는 message-passing이나 3D convolutional neural network 등을 이용해 protein backbone을 입력으로 받아 structural feature를 생성하고, 그로부터 최적의 a.a. sequence들을 decoding 해주는 encoder-decoder 구조를 사용한다. 또한 Chorma나 RFdiffusion 같은 denoising diffusion probabilistic models에 기반한 모델들은 gaussian noise로부터 새로운 단백질 서열을 생성하거나, 특정한 구조에 제약을 둔 형태의 서열들을 생성할 수 있다.
위와 같이 DNNs, GNNs (Graph neural networks; e.g. message-passing)과 풍부한 데이터로 인해 de novo protein design의 진보가 일어나고 있는 상황에서, 본 논문은 아래와 같은 어려움을 제시한다.
While powerful in generating medium to large novel protein folds, many DNNs encouter challenges in generating small new protein domains not observed in nature, i.e., domains with novel configurations or wiring of the SSEs.
즉, 자연에 존재하지 않는 작은 단백질의 domain을 생성할 때, model이 생성하는 backbone들이 결과적으로 novel하기 보다는 PDB에 존재하는 단백질들의 일정 부분 이상 모방이 되는 경우가 많다는 것이다 (물론 medium, large protein에 대한 generability에 관련된 참고문헌이 제시되어 있지만, 아직 확인해보지는 않았다). 실제로 나 역시 최근 trasformer 구조에 기반한 multi modal generative model인 ESM3를 이용해서 active site의 서열과 구조만을 고정하고 de novo로 단백질의 서열을 생성하는 작업들을 해보고 있는데, 생성 과정의 sharpness를 조절하는 temperature에 많은 조작을 가해도 생성되는 서열들의 reference 서열에 대한 유사도가 40~60% 정도로 유지되어서 어느 정도 generability 상의 한계점이 있는 것 같다는 생각을 해보았었다. 최근 활발하게 연구되는 AI-based drug discovery에서는 처리하고자 하는 물질에 맞는 target structure를 만들고, 그로부터 서열을 생성하게 될텐데, 만약 target structure가 모델이 학습하지 않은 novel한 구조일 경우 그로부터 잘 작동하는 서열을 만드는 데에 지금 논의하고 있는 문제도 중요한 요소가 될 것이라는 생각도 든다.
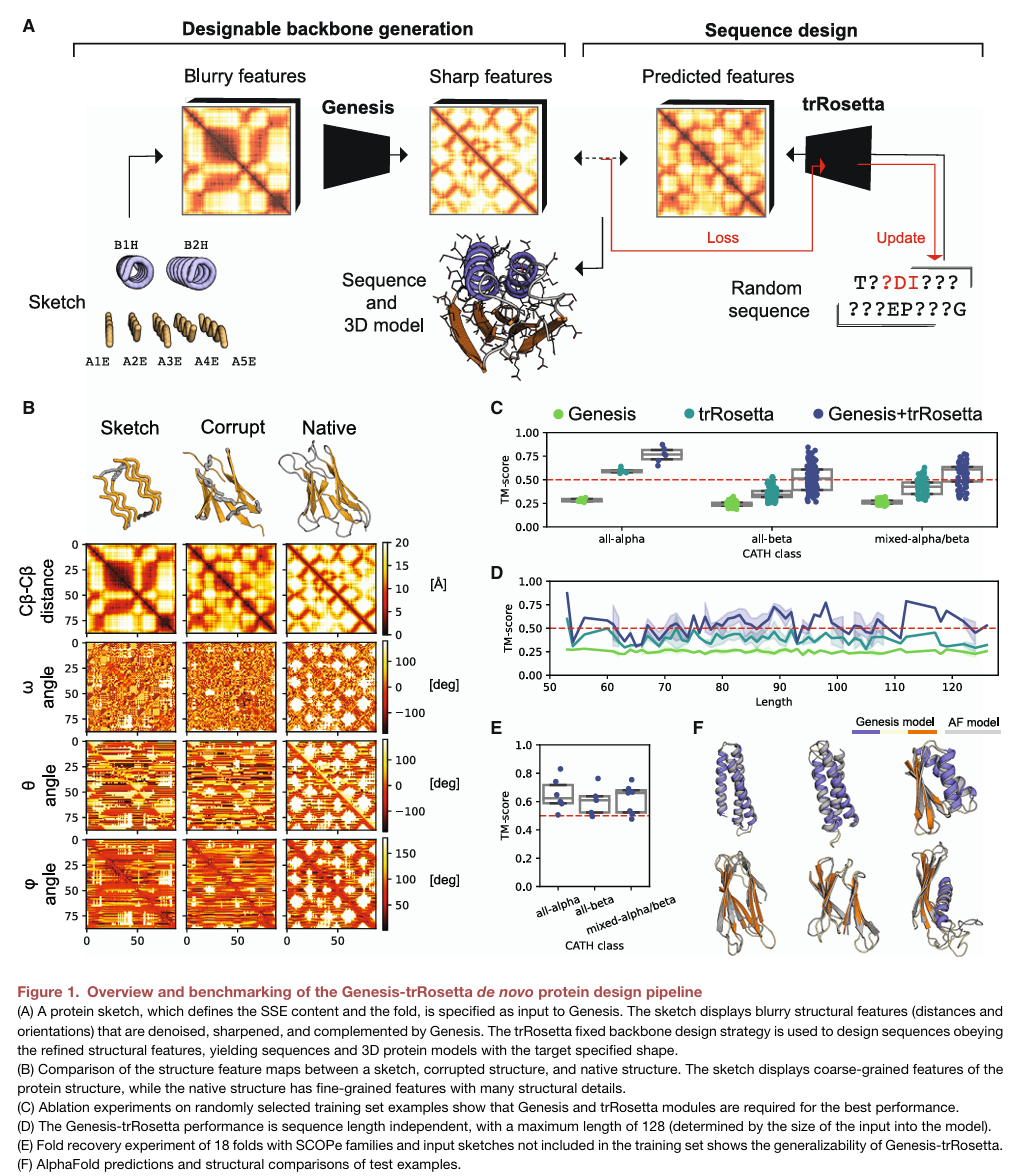
결과적으로 저자들은 small novel protein folds를 designing하는 어려움을 분석하고, protein backbone으로부터 3D structural pattern을 학습하는 새로운 접근법인 backbone designibility를 제안하였다. 전체적인 구조는 다음과 같다.
1) 먼저 protein fold의 representation을 string format으로 준비하고, 이를 sketch (3D low-resolution representation of the fold where SSEs are placed onto layers; SSE (secondary structural elements))라는 형태로 3D space로 projection 한다.
2) 이때 sketch의 designability를 증가시키기 위해서 Genesis (a convolutional variational autoencoder; convolutional VAE)를 거쳐서 sketch의 distance와 orientation을 샘플링이 가능한 compact latent representation으로 encoding하고, 이후 native-like distance & orientation probabilities로 decoding한다.
3) Refined distance & orientation probabilities를 target template로 사용해서 trRosetta가 새로운 backbone과 sequence들을 template restraints에 맞추어 생성하도록 유도한다.
저자들은 위와 같은 pipeline을 이용해서 5 native folds & 3 darkfolds (with different secondary structure and loop lengths)로부터 서열을 생성하고, protease resistance로부터 550 designs의 folding을 평가하였다.
Results and Discussion
A computational pipeline for enhanced backbone designability
- Variational Autoencoder (VAE): Variational autoencoder는 기본적으로 input x를 받아 x를 condition으로 이용하는 conditional distribution으로 정의되는 encoder $q(z\mid x)$ 로부터 latent variable z를 샘플링하여 얻고, 다시 z를 condition으로 이용해서 원래의 x를 복원하는 decoder $p(x\mid z)$ 를 구성하여 최종적으로 x’ 를 얻어 input을 복원할 수 있도록 학습하는 architecture이다.
이들이 제안한 de novo design workflow의 핵심이 되는 deep learning module은 Genesis라는 module로, 앞서 언급했듯 convolutional VAE에 기반하고 있고, low-resolution sketches에 native-like structural feature를 부여하여 sharp feature를 만들어주는 역할이다. 내가 이해한 바로는, pre-training 과정에서 native fold의 feature를 복원하도록 encoder/decoder를 학습하고, 이후 다량의 sketches & native fold pair를 이용해서 fine tuning하여 sketches를 input으로 주면 그의 native-like structural feature를 출력하도록 한 것이다. 그리고 결국 Genesis의 output은 trRosetta 같은 모듈이 사용 가능한 backbone을 제공하는 것이기 때문에, 실제 학습 시에 fold에서 loop 부분은 corruption을 시켜 학습할 것으로 보인다. 실제 추론 과정에서 Genesis에 들어가는 input은 이동이나 회전에 invariant하도록 atomic coordinate가 아닌, pairwise distance & orientation을 이용한다. 즉, 과정을 정리하면
1) sketch에서 실수 값의 pairwise distance & orientation map (Blurry features)을 얻고
2) map을 condition으로 이용해서 latent를 얻은 후
3) 그로부터 native-like conformation map (Sharp features)을 얻어
4) trRosetta를 이용해 guided sequence search를 진행
참고로, 인공지능 논문에도 매번 등장하는 ablation study를 randomly selected 273 folds에 대해 수행했을 때, Genesis, trRosetta, both에서 나온 결과를 원래의 구조와 aligning 해보는 self-consistency TM score를 보면 Genesis-trRosetta를 사용하는 protocol이 전반적인 성능이 가장 좋다는 것을 확인할 수 있다. 추가로 아래 figure 1.E-F를 살펴보면, 이 framework가 traning set에 포함되지 않은 SCOPe (Structural Classification of Proteins-extented) family에 대해서도 일반화된 좋은 예측을 보여주고 있음을 확인 가능하다.
다만, 이러한 방법론에서도 $\beta$-helix folds, $\beta$-Prisms와 같이 같은 수의 $\beta$ sheet를 가지는 것처럼 비슷한 형태인 경우, sketch가 잘 구별되지 않아서 결과적으로 TM score가 낮게 나오는 경우가 많다는 것을 언급하고 있는데, 만약 내가 나중에 비슷한 형태의 경량화된 모델이나 architecture를 설계할 때 고려해야 할 breakthrough가 필요한 부분으로써 함께 정리해보았다.
Large-scale de novo design of native topologies
본격적으로 Genesis-trRosetta de novo design framework의 성능을 보기 위해서, 저자들은 5개의 다른 topologies를 design하는 것으로 시작하였다 (ubiquitin-like, Rossmann fold, immunoglobulin-like, jellyroll fold, and Top7 fold). 이들 중 Top 7의 경우 de novo로 합성된 구조이지만, 어느정도 natural repertoire를 띄고 있다. 그리고 fold designability를 표시하기 위해 relative contact order라는 개념을 사용하였다. 아래의 개념을 참고해서, lg-like와 jellyroll folds의 경우 contact order가 ~0.20 & 0.26 정도로 나머지 folds에 비해 높아서 structural contacts의 non-locality가 높고, 그만큼 design이 어렵다는 것을 알 수 있다.
Contact order: 단백질의 folded structure 상에서 접하는 residue들 간의 sequence 상의 평균 거리. Contact order가 클수록 local한 contact을 통해 접히지 않고 folding에 시간이 소요되므로, design의 난이도도 높아짐.
design 방식 (exploration-exploitation)은 다음과 같다.
1) Prior knowledge가 없기 때문에, 20-30개의 secondary structure + loop length combinations을 제작 (candidate search)하고 TM score와 wasserstein distance를 이용해서 filtering
2) 앞선 단계에서 생성된 20,000개 정도의 서열들과 3차 구조 모델을 AlphaFold의 pLDDT score와 TM score (between Genesis & AlphaFold)를 이용하여 filtering (production stage; figure 2-A)
3) 각 fold에서 50개의 design set을 선정하여, antibody를 통해 protease (trypsin, chymotrypsin)를 내는 yeast에 protein을 붙히고, digestion에 대한 저항성을 확인
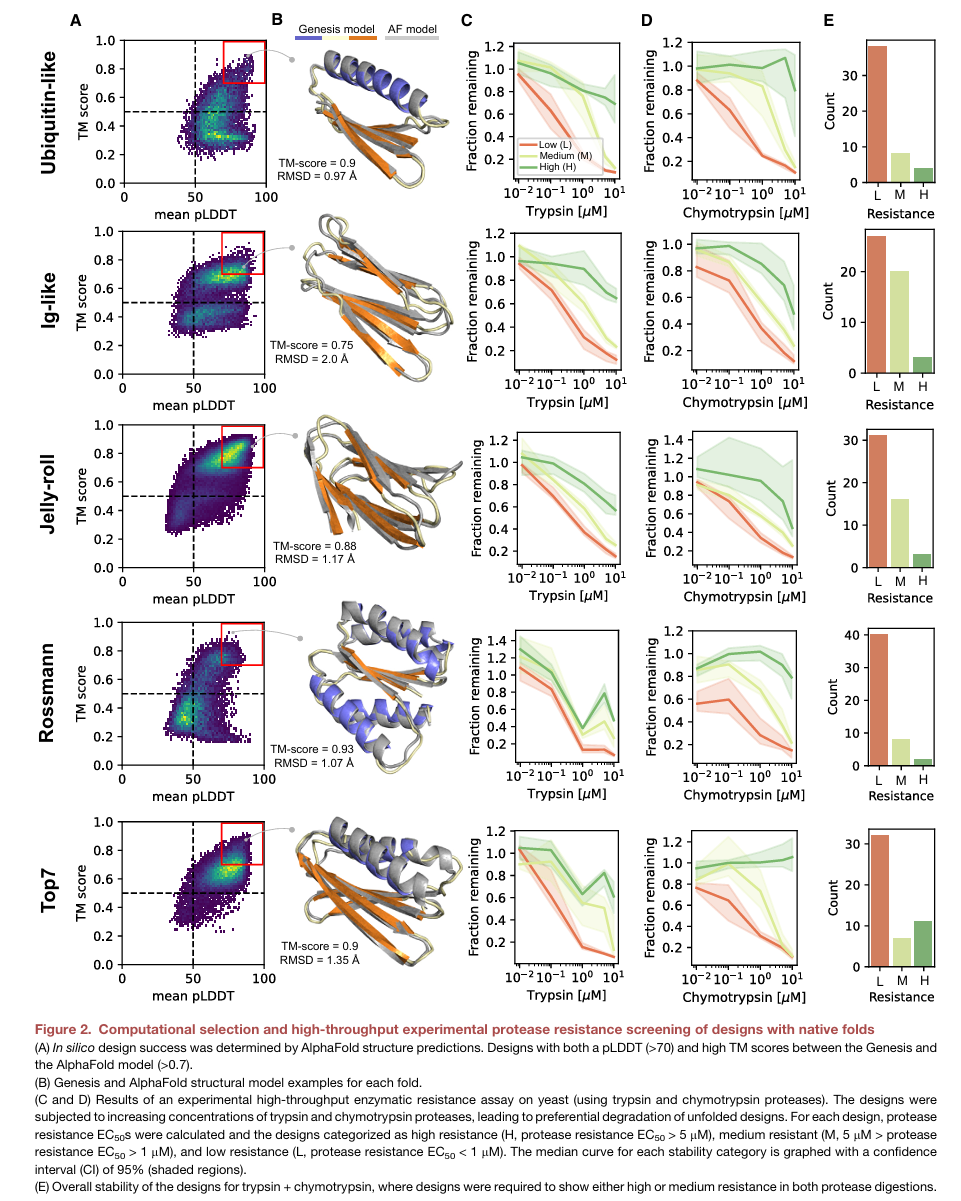
결과적으로 5 종류의 fold에 대해서 10% 정도는 highly stable한 design들이 나왔고, 그 비율이 일반적으로 어렵다고 알려진 jellyroll, ig-like fold에서도 유지되는 것을 보아, 저자들은 framework이 잘 작동한다는 것을 보였다.
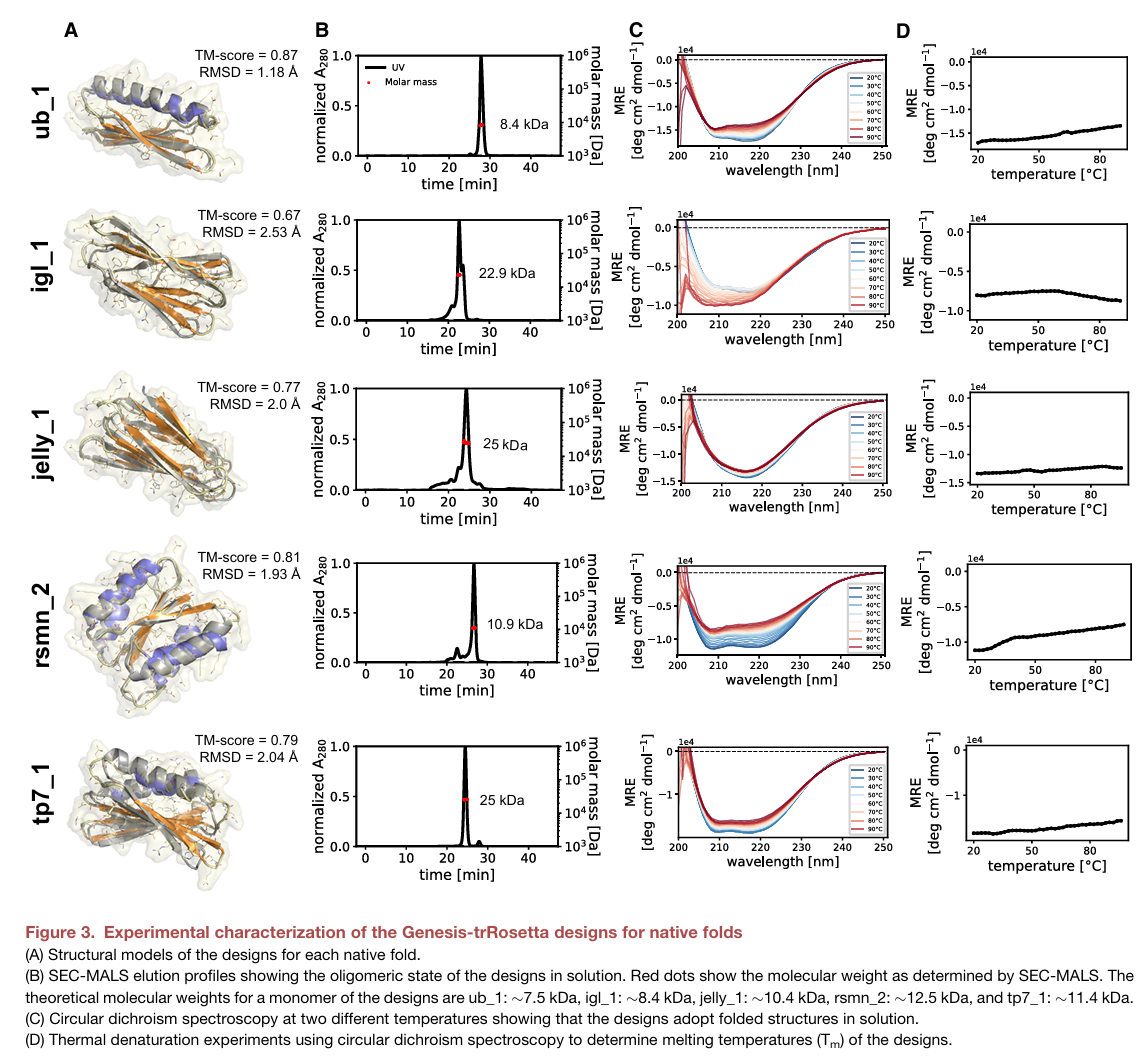
연속적으로 저자들은 앞선 design들 중에서 medium or highly digestion-resistant designs를 뽑아서 biochemically characterize를 실시했다. 논문에서는 몇 개의 design을 선택하고, 각각에 대한 분석결과를 포함하고 있지만, 나는 figure 3에 대해서 어떤 분석법을 적용한 것이며, 왜 했는지에 대해 정리해보았다.
아래의 figure 3에서 3-B는 SEC-MALS (Size-Exclusion Chromatography - Multi-Angle Light Scattering)의 결과로 peak의 수와 분자량 등을 확인하기 위해 확인하는 것이다. 5 종류의 fold의 대표 design들에서 모두 peak의 분포가 하나인 것을 확인할 수 있다 (monodisperse). 다만, ub_1의 경우 이론적인 질량 (~7.5kDa)과 측정한 질량이 비슷하지만, igl_1의 경우 2배 이상 차이 나는 것을 보아 oligomer 형태로 존재하는 것을 예상할 수 있다. Figure 3-C는 circular dichroism spectroscopy의 결과로 secondary structure에 민감하게 작동하는 분광현미경의 결과인데, 각 온도 변화에 대해 점진적이고 비슷한 패턴을 보이는 것을 통해 solution 상에서 folding을 잘 이루고 있다는 것을 확인할 수 있다. 마지막 3-D는 동일한 spectroscopy로 melting temperature를 확인하는 것이다.
최종적으로 product stage를 거쳐 20,000개 정도의 후보 서열들로부터 protease resistance를 보이고, purified and biochemically characterized된 protein들은 10개 정도 된다는 것에서 결국 validation이 정말 중요하다는 것을 다시 한 번 확인할 수 있었다.
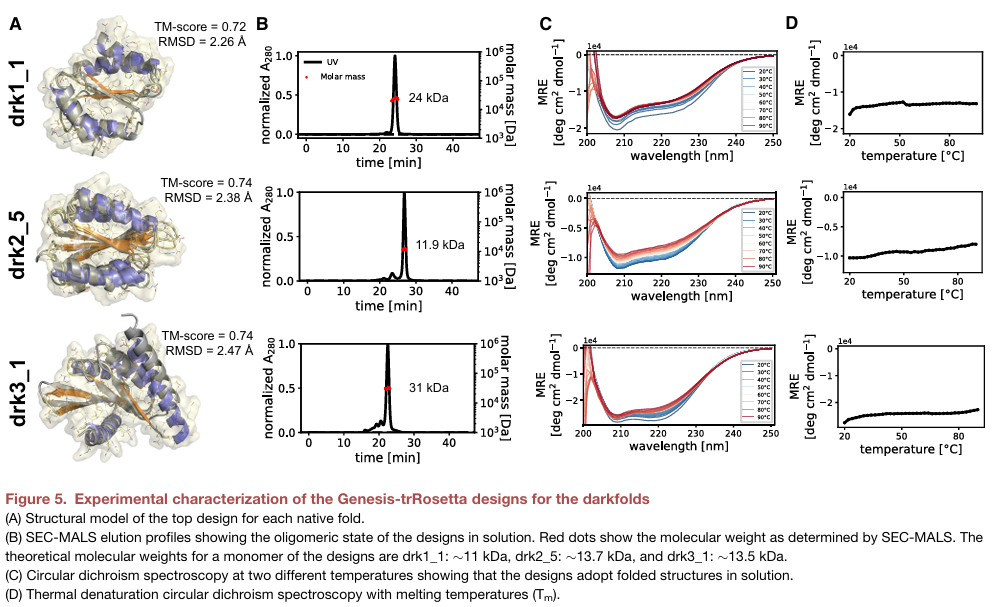
Exploration of “dark-matter” folds
de novo protein design의 궁극적인 목표 중 하나는 new functionalities에 적용이 가능하도록 natural repetoire를 넘어서서 존재하는 protein fold에 대해 design을 하는 것이다. 즉, framework trained on native protein’s structural data is capable of generalizing outside the distribution of natural folds? 라는 질문에 대한 답을 찾는 것이다.
저자들은 이에 대해 답하기 위해서 training set에 존재하지 않으며, natural repetoire에도 존재하지 않는 folds를 최대한 샘플링 하였다. 그 방식은 기본적으로 Talyor와 collegues에 의한 선행 연구를 활용하는 것이다. Three-layer mixed-$\alpha/\beta$ fold space 중에서 탐색되지 않은 부분을 사용하되 C$\alpha$-trace를 이용해서 SSE의 handedness나 loop connectivity 같은 최소한의 natural한 protein structure를 따르고, 추가로 그들 중 동일 layer 상에 secondary structure type이 섞였거나, SSE가 제 기능을 하지 않거나, loosely packing, and crossing loop의 경우를 제거하여 줄인 후 3가지의 distinct folds를 선택하였다.
만약 내가 dark fold space에 대한 generalizability의 문제를 다루게 된다면, 이후 dark fold를 생성한 방법에 대해서는 구체적으로 더 공부해야 할 것 같다.
아무튼 natural folds와 동일한 과정의 분석 결과를 살펴보면, candidate progress에서 얻은 novel folds에 대한 대부분의 design들이 median TM score가 0.5를 넘었고, median RMSD with AF prediction이 3.7$\,\text{\AA}$ 정도로, AF의 예측과 꽤 잘 맞아들어가는 것을 확인할 수 있었다. 최종적으로 74개의 candidate에 대해서 protease digestion assay를 진행하였고,

아래의 Figure 5에서는, best designs에 대한 purification과 biochemical characterization의 결과를 보여준다. 논문의 supplementary에는 좀 더 많은 design들에 대한 결과를 포함하고 있는데, 꽤 많은 design에서 설계한 sketch에 맞는 CD spectrometry 결과를 보이면서, 높은 thermostability를 가지고 있는 것을 확인할 수 있었다. 즉, 이 결과들로부터 Genesis-trRosetta framework가 학습에서 포함되지 않은 novel folds에 대해 일반화되며, nature에서 발견되지 않은 folds에 대해서도 backbone generation과 sequence search가 가능하도록 구조적인 패턴을 학습했다는 것을 제시하고 있다.
Mapping successful designs onto the current protein sequence and fold space
- CATH database: 단백질 구조의 domains를 3 classes로 구분 (all-$\alpha$, all-$\beta$, mixed-$\alpha/\beta$). 이 classes들은 SSEs의 배열에 따라 40개의 unique architecture로 구분, architecture들은 SSE connectivity에 따라 1291개의 folds로 구분.
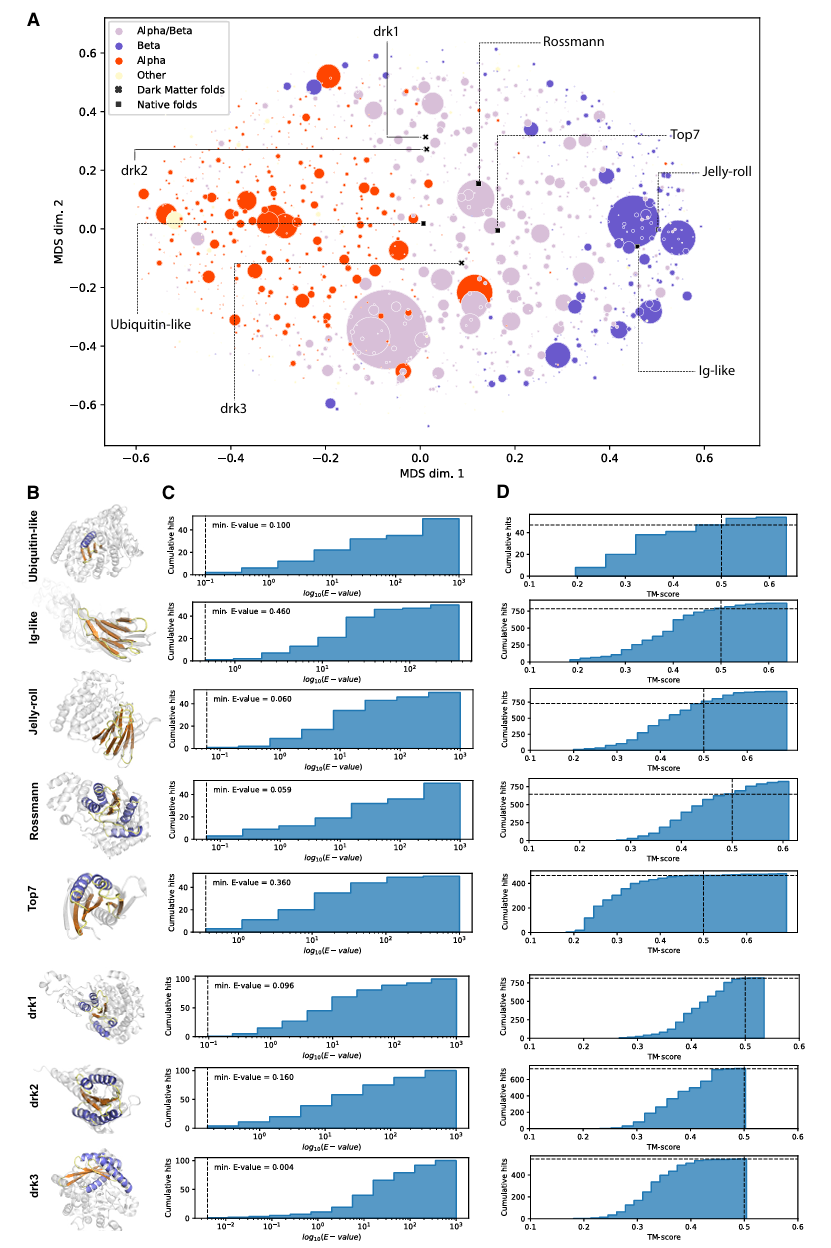
Designed protein sequences들이 알려진 protein domain에 대해 얼마나 구조적으로 유사한지를 확인하기 위해서, 본 연구에서 얻은 native & darkfold 각각의 representative model이 모든 CATH domain representative와 TM-align algorithm을 통해 aligned 되었다. 그 결과로 나온 TM score similarity matrix가 classical multidimensional scaling에 의해 2D plot으로 mapping된 결과가 아래의 Figure 6-A이다. 그림을 살펴보면, nature folds와 달리 dark folds의 경우 CATH domain에 속하지 않고 분리되어 있는 것을 확인할 수 있다.
Sequence novelty의 측면에서, protein 서열의 유사도를 비교할 수 있는 BLASTp를 이용해서 non-redundant database (NR)에 대해 비교했을 때 유의하게 비슷하면 0.001 이하의 값을 보여야 하는 E-value가 전부 0.001 이상으로 나왔다. 즉, naturally occuring sequence들과는 차이가 존재한다는 것을 알 수 있다. 또 structure novelty의 측면에서, fast FoldSeek algorithm을 이용해서 PDB의 structural hit를 살펴봤을 때, 3 종류의 darkfolds에서 모두 TM score가 0.5를 넘는 경우가 거의 없었다.
이후 저자들은 각 darkfold에 대해 PDB, CATH, AF DB에서 가장 맞는 matching을 찾고, 그로부터 novel fold의 기원이나 특징 등에 대한 생각들도 서술하고 있다. 재밌는 점은 이들의 backbone을 tertiary motif로 분해해서 분석하기 위해 TERMs analysis method를 적용했다. TERMs analysis는 protein backbone을 tertiary motif로 쪼개어, NR (non-redundant) database에서 얼마나 자주 등장하는 지를 통해서 protein backbone의 designability score를 계산할 수 있다. 이 방식을 통해 drk1~drk3의 folds를 분석해보면, 각각 motifs의 abundance가 다를 뿐 아니라 dark matter protein 마다 C-terminus, N-terminus 같이 natrual folds에서 잘 등장하지 않는 특이한 구조를 가진 위치도 다 다르다는 점을 확인할 수 있었다. 즉, darkfolds 자체는 nature에서 novel하지만, 그들의 structural fragments는 자연에서 이미 많이 사용되고 있고, 그 조합이나 정도의 특이성이 존재한다는 것이다.
Comment
개인적으로 protein sequence & fold space라는 것에 대해 개인적으로 관심이 있었고, 인공지능이 가지고 있는 학습 데이터 너머의 일반화 가능성에 대해서도 궁금한 점이 있었어서 나름 재밌게 논문을 읽었던 것 같다. 이런 방법들이 사용될 수 있는 곳을 생각해봤을 때, 예를 들어 신약 개발을 위해 특정한 구조의 항체를 만들고 싶다면, Genesis-trRosetta의 arichitecture를 활용해서 자연에 존재하지 않는 조합의 folds에 대해서도 적절한 서열을 많이 생성하고, 좋은 design들을 얻어낼 수 있을 거라는 기대가 된다. 다음 번에는, 좀 더 sequence & fold space의 근간이 되는 논문을 읽어볼까 하는데, 그로부터도 많은 것들을 배울 수 있을 것 같다.